
edgeR基因表达差异分析
文章目录
官方文档总结
注意⚠️:
- edgeR设计用于实际读取计数。我们不建议将预测的转录本丰度输入到edgeR而不是实际计数中。
读取read数
可以使用read.table(),read.delim(),如果文件比较多,readDGE(files, columns=)来一次性读取多个文件。注意,readDGE()需要指定两列,一列用于计数,一列用于基因标识符
x <- readDGE(files, columns=c(1,3)) DGEList对象、构建分组
DGEList是一个可以包含多种内容和统计的列表。DGEList至少需要的元素:counts、samples(包含group分组信息和lib.size文库大小),counts用来存放表达矩阵,samples用来标记样本信息和库的大小,group声明组别 。
# 构建一个含有组别标记的DGEList y<- DGEList(counts=x) group <- c(1,1,2,2) y<- DGEList(counts=x,group=group) 过滤,删除低表达基因
低表达基因不仅用不到,而且会干扰结果,所以要去除在任何样本中都没有足够多的序列片段的基因应该从下游分析中过滤掉,因为:
- 低表达没有生物学意义
- 去除低表达数据可以对数据中均值-方差关系有更精确的估计
- 减少了观察差异表达下游分析中的运算量
edgeR包中的filterByExpr函数提供了自动过滤基因的方法,可保留尽可能多的有足够表达计数的基因。此函数默认选取最小的组内的样本数量为最小样本数,保留至少在这个数量的样本中有10个或更多序列片段计数的基因。
过滤标准是,以最小对组内样本数为标准,(此例最小组内样本为3),如果有基因在所有样本中表达数(count)小于10的个数超过最小组内样本数,就剔除该基因。换算为cpm即cut.off.cpm=10/
CPM标度转换
cpm<- edgeR::cpm(x) lcpm <- edgeR::cpm(x, log=TRUE) 手动过滤
经验设置为cpm=1位为cutoff点。但是,这并不是最精准。因为随着测序深度增加,例如20million(2 千万),cpm=1 意味着 counts=20。阈值可能会有点高。测序深度低的话,例如2million(2百万),cpm=1 意味着counts=1。阈值可能会太低。此时可以使用自动过滤,或者根据cut.off.cpm=10/
来计算。
cpm(y) keep_cpm <- rowSums(cpm(y)>1) >= 3 #此例设置最小组内样本数为3 x<- x[keep_cpm,,keep.lib.sizes=FALSE] 自动过滤
使用edgeR::filterByExpr()自动过滤
keep.exprs<- edgeR::filterByExpr(x,group=group) x<- x[keep.exprs,,keep.lib.sizes=FALSE] 归一化
测序深度
不同文库大小代表不同测序深度。这是基本建模过程的一部分,并自动进入倍数变化或p值计算。它始终存在,不需要任何用户干预。
有效库大小
y <- calcNormFactors(y) 注意⚠️:
归一化并不会直接在counts数值上修改,而是归一化系数会被自动存在x$samples$norm.factors。
GC含量
基因长度
因为GC含量和基因长度在每个基因间不会改变,因此这些都是相对的,因此对差异分析影响很小。
MDS图形展示 样本无监督聚类
这种图表使用无监督聚类方法展示出了样品间的相似性和不相似性,能让我们在进行正式的检验之前对于能检测到多少差异表达基因有个大致概念。
# 图形展示 library(RColorBrewer) lcpm <- cpm(y, log=TRUE) col.group <- group levels(col.group) <- brewer.pal(nlevels(col.group), "Set1") col.group <- as.character(col.group) plotMDS(lcpm,labels=group,col=col.group) #或者直接使用 plotMDS(y) 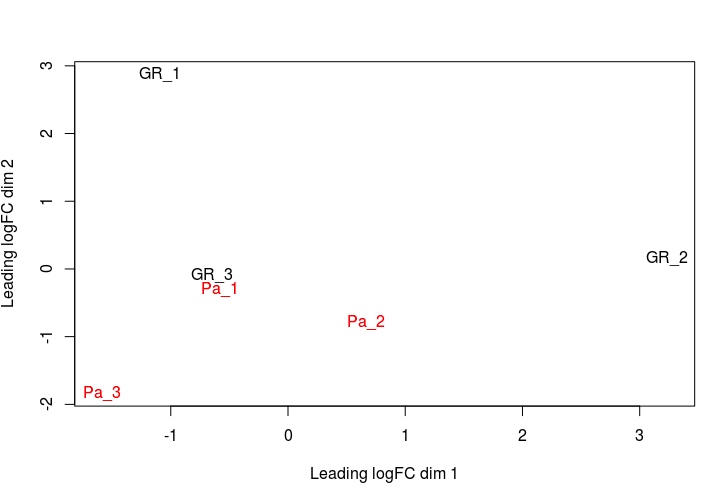

负二项式模型
计算生物变异系数
y < -estimateDisp(y) 或者先计算common离散度,再计算tagwise离散度
y <- estimateCommonDisp(y) y <- estimateTagwiseDisp(y) 计算差异基因
et<- exactTest(y) topTags(et) 广义线性模型(Glm)
对于更复杂的实验设计(有多个因素),可以通过广义线性模型来及性能你和。
计算离散度
通过下面来估计common离散度、trended离散度、tagwise离散度。
y<- estimateDisp(y,design) 或者分开依次进行
y <- estimateGLMCommonDisp(y, design) y <- estimateGLMTrendedDisp(y, design) #估计trended离散度 y <- estimateGLMTagwiseDisp(y, design) 关于design的构建(?):
#group是区别分组的factor对应不同样本,lane是不同样本的的lane道 design <- model.matrix(~0+group+lane) colnames(design) <- gsub("group", "", colnames(design)) 计算DE基因
# 例子 group <- factor(c(1,1,2,2,3,3)) design <- model.matrix(~group) fit <- glmQLFit(y, design) 如果没有重复样本?
首先确定一个良好的BCV,人类数据一般设定为0.4
一个例子:
bcv <- 0.2 counts <- matrix( rnbinom(40,size=1/bcv^2,mu=10), 20,2) y <- DGEList(counts=counts, group=1:2) et <- exactTest(y, dispersion=bcv^2) 如果是人类数据:
y_bcv <- y bcv <- 0.4 et <- exactTest(y_bcv,dispersion = bcv ^2) 输出结果
result = topTags(et, n = nrow(et$table))$table 查看统计
summary(de <- decideTestsDGE(et)) 我想建立并管理一个高质量的生信&统计相关的微信讨论群,如果你想参与讨论,可以添加我微信: veryqun 。我会拉你进群,当然有问题也可以微信咨询我。
对我唯一的精神鼓励可能就是下方的点赞了吧 ^ ^
发布者:全栈程序员-站长,转载请注明出处:https://javaforall.net/209219.html原文链接:https://javaforall.net


