
大家好,又见面了,我是你们的朋友全栈君。
平日经常会分析shotgun宏基因组的数据,我们的pipeline使用MetaPhlAn,Kraken等profiler。这种数据经常会产生一个表格,如下
download.file("https://bitbucket.org/biobakery/biobakery/raw/tip/demos/biobakery_demos/data/metaphlan2/output/SRS014459-Stool_profile.txt", 'SRS014459-Stool_profile.txt')
knitr::kable(head(read.table('SRS014459-Stool_profile.txt')))| V1 | V2 |
|---|---|
| k__Bacteria | 100.00000 |
| kBacteria|pFirmicutes | 64.91753 |
| kBacteria|pBacteroidetes | 35.08247 |
| kBacteria|pFirmicutes|c__Clostridia | 64.91753 |
| kBacteria|pBacteroidetes|c__Bacteroidia | 35.08247 |
| kBacteria|pFirmicutes|cClostridia|oClostridiales | 64.91753 |
第一列是分类信息注释,第二列是相对丰度(百分比)。在做这种图可视化方面,目前个人见过最强大的是GraPhlAn:
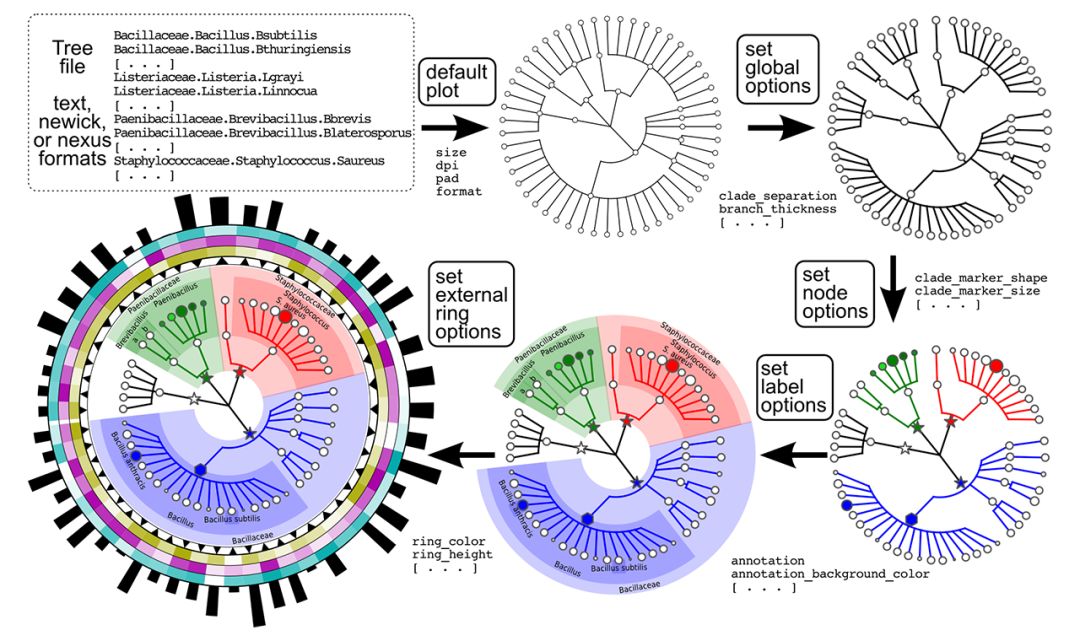

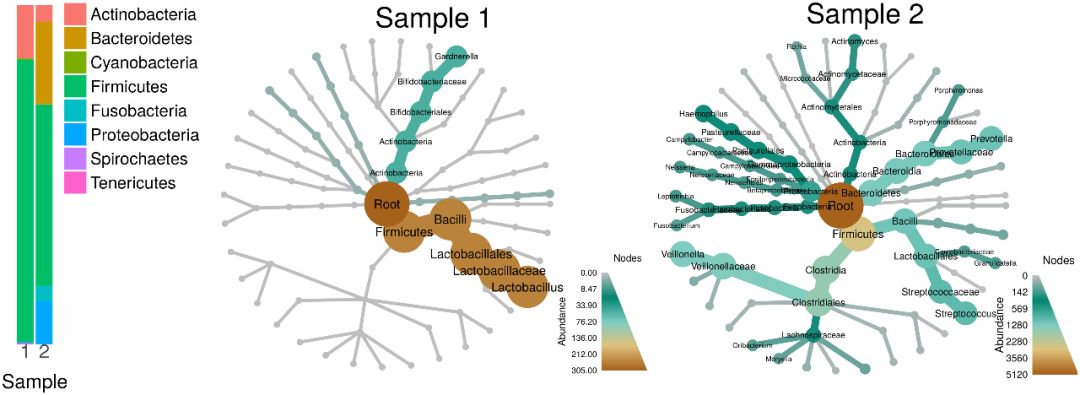
官网上相关的教程很详细,但是问题是,这个完全封闭的python程序,想要hack,还真的是挺难得。Krona可能是另一个选择,但是同样还是会有同样的问题。最近发布的R包Metacoder,画出的图个人真心不是很喜欢:
跟Y叔讨论了一下用ggtree实现像GraPhlAn那样图的可能性,得到了肯定的答复,于是开始自己造轮子。
MicrobiomeViz–千里之行,始于足下
其实可以写一个简单的函数,但是还是想做一个拓展性更强的东西,所以就有了这个包(不断完善中): https://github.com/lch14forever/microbiomeViz
使用实战
让我们产生lefse调用graphlan绘制的物种树标记差异物种的Cladogram
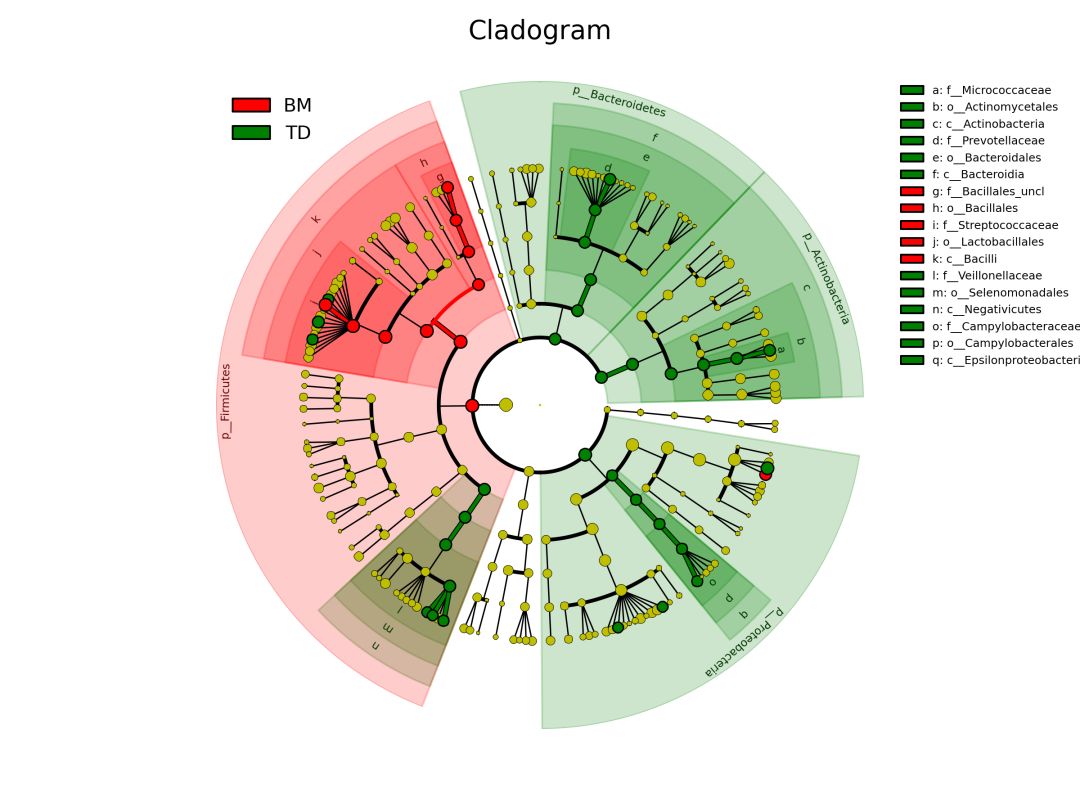
输入数据为metaphlan2结果合并的矩阵。如何生成详见:MetaPhlAn2一条命令获得宏基因组物种组成
ID BM_SRS013506 BM_SRS015374 BM_SRS015646 BM_SRS017687 BM_SRS019221 BM_SRS019329 BM_SRS020336 BM_SRS022145 BM_SRS022532
k__Bacteria 100.0 100.0 100.0 100.0 100.0 100.0 100.0 100.0 100.0 100.0 100.0 100.0 100.0 100.0 100.0 100.0 100.0
k__Bacteria|p__Actinobacteria 1.33609 2.90435 0.45117 6.7964 14.08966 2.30709 7.30108 0.53534 3.57207 8.47622 7.07037 17.30722 30.62601
k__Bacteria|p__Actinobacteria|c__Actinobacteria 1.33609 2.90435 0.45117 6.7964 14.08966 2.30709 7.30108 0.53534 3.57207 8.47622 7.07037 17.30722包安装和加载
# microbiomeViz需要 R 3.5 以上,依赖包安装
source("https://bioconductor.org/biocLite.R")
biocLite("ggtree")
devtools::install_github("lch14forever/microbiomeViz")
library(microbiomeViz)物种相对丰对矩阵绘制物种树
# 加载测试数据
df <- read.table("http://bailab.genetics.ac.cn/markdown/R/microbiomeViz/merged_abundance_table.txt", head=TRUE, stringsAsFactors = FALSE)
## 计算均值用于呈现结点大小
dat <- data.frame(V1=df[,1], V2=rowMeans(df[,-1]), stringsAsFactors = FALSE)
# 用物种和丰度生成树骨架
tr <- parseMetaphlanTSV(dat, node.size.offset=2, node.size.scale=0.8)
p <- tree.backbone(tr, size=0.5)
p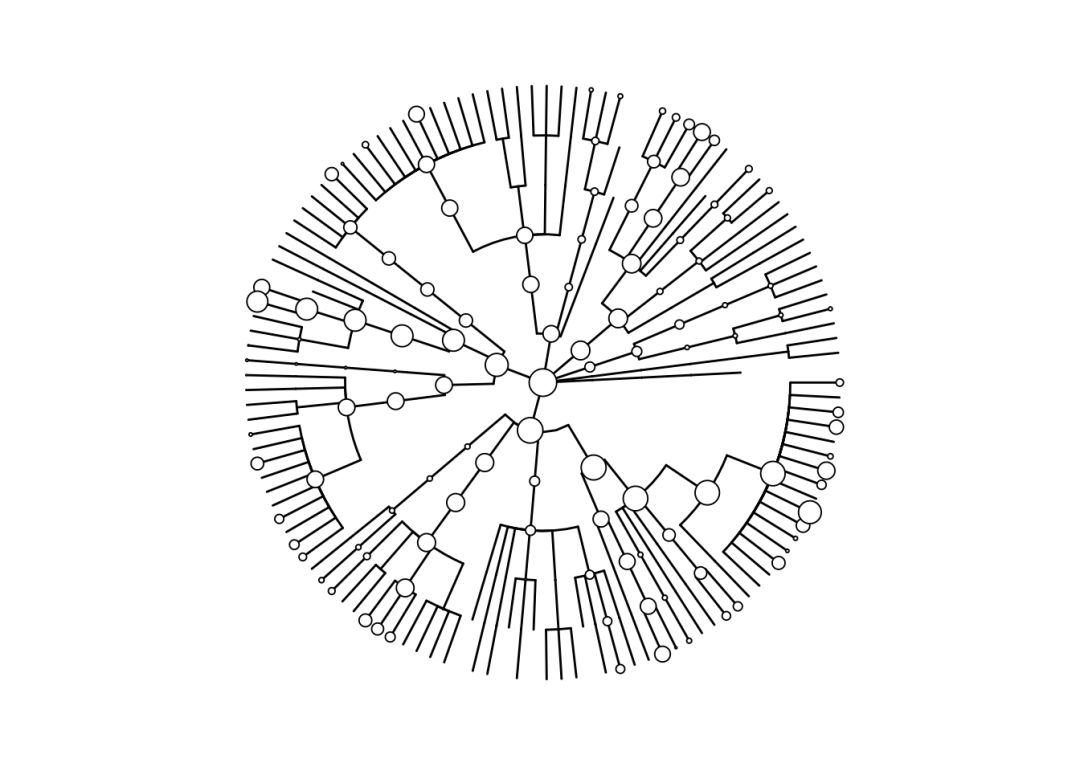
差异物种注释
# 读取需要颜色标注的差异物种列表,本质上是两列和颜色对应表
lefse_lists = data.frame(node=c('s__Haemophilus_parainfluenzae','p__Proteobacteria',
'f__Veillonellaceae','o__Selenomonadales',
'c__Negativicutes', 's__Streptococcus_parasanguinis',
'p__Firmicutes','f__Streptococcaceae',
'g__Streptococcus','o__Lactobacillales',
'c__Bacilli','s__Streptococcus_mitis'),
color=c(rep('darkgreen',6), rep('red','6')),
stringsAsFactors = FALSE
)
# 注释树
p <- clade.anno(p, lefse_lists, alpha=0.3)
p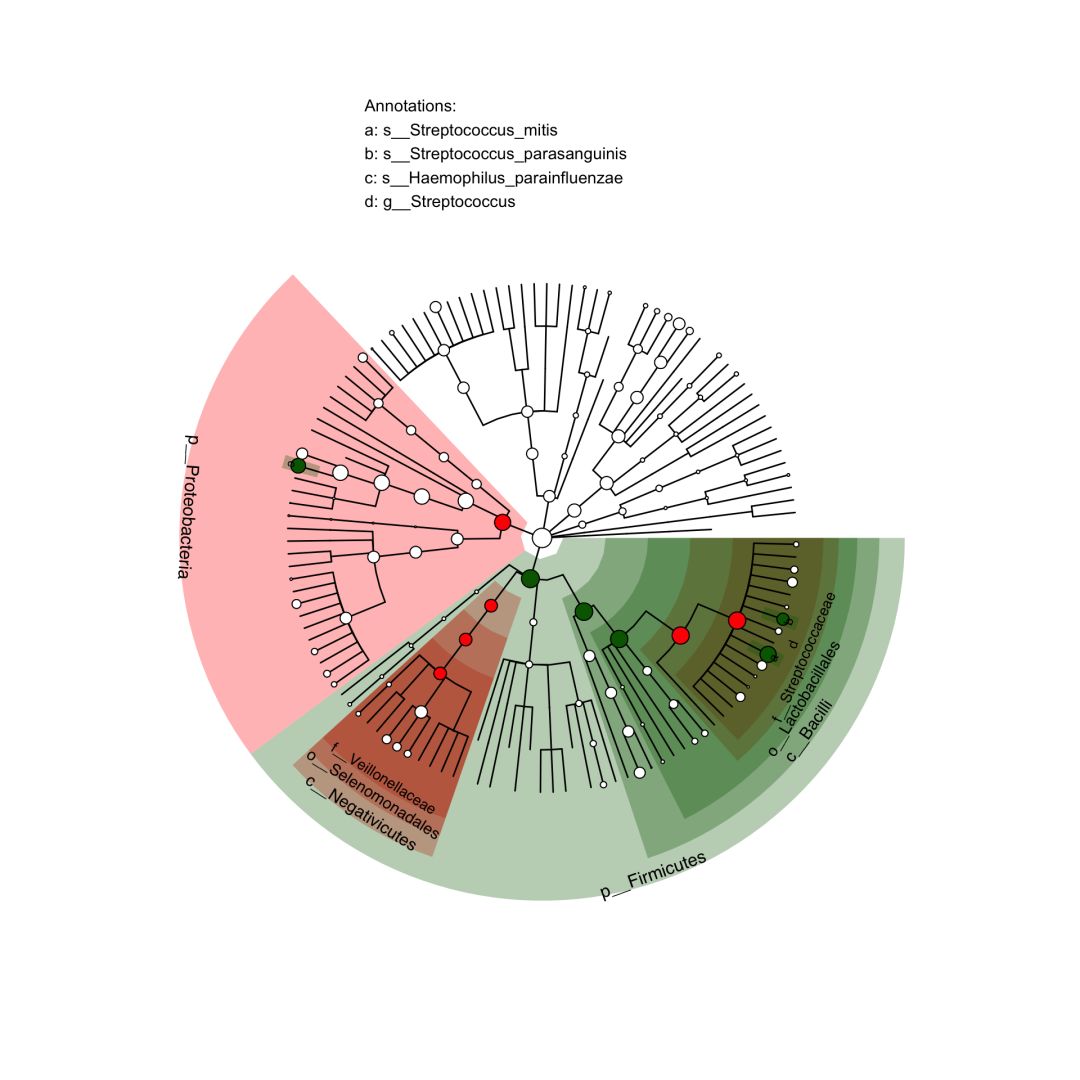
简单几行代码,美图大功告成。
Reference
http://lchblogs.netlify.com/post/2018-01-18-r-metagenomeviz/
http://lchblogs.netlify.com/post/2018-04-20-r-microbiomeviz_example/
发布者:全栈程序员-站长,转载请注明出处:https://javaforall.net/144822.html原文链接:https://javaforall.net


