
软件介绍
基因组选择中, 有时候测量了很多家系,如果想看一下这些家系的分类情况,可以使用软件对其进行分群。一般使用的软件就是STRUCTURE,但是STREUTURE运行速度极慢,admixture凭借其运算速度,成为了主流的分析软件。下面介绍一下admixture的使用方法。
官方网址
Admixture
http://software.genetics.ucla.edu/admixture/download.html


软件安装
使用conda进行软件安装.
conda install admixture 安装完成之后, 键入admixture, 显示如下信息, 说明安装成功
(base) [dengfei@localhost test]$ admixture ADMIXTURE Version 1.3.0 Copyright 2008-2015 David Alexander, Suyash Shringarpure, John Novembre, Ken Lange Please cite our paper! Information at www.genetics.ucla.edu/software/admixture Usage: admixture
See --help or manual for more advanced usage.
目录

1. 快速起步
1.1 下载示例数据
注意,官网上的示例数据已经无法下载,想要测试数据,可以关注公众号:“育种数据分析之放飞自我”,回复“admixture”,获得测试数据。——–2020-5-23 更新
wget http://software.genetics.ucla.edu/admixture/hapmap3-files.tar.gz 下载完成之后, 解压:
tar zxvf hapmap3-files.tar.gz 查看解压后的文件:
(base) [dengfei@localhost admixture]$ ls hapmap3.bed hapmap3.bim hapmap3.fam hapmap3-files.tar.gz hapmap3.map 1.2 admixture支持的格式
- plink的bed文件或者ped文件
- EIGENSTRAT软件的
.geno格式
注意: - 如果你的数据格式是plink的bed文件, 比如
a.bed, 那么你应该包含a.bim,a.fam - 如果你的数据格式是plink的ped文件, 比如
b.ped, 那么你应该包括b.map
1.3 选择合适的分群数目k值
这里你要有一个k值, 如果你不知道你的群体能分为几个类群, 可以做一个测试, 比如从1~7分别分群, 然后看他们的cv值哪个小, 用那个k值.
1.4 运行k=3的admixture
注意, 这里的名称为hapmap3.bed, 而不是hapmap3(不像plink那样不加后缀), 而且没有--file 参数, 直接加plink的bed文件
admixture hapmap3.bed 3 运算结果:
(base) [dengfei@localhost admixture]$ admixture hapmap3.bed 3 ADMIXTURE Version 1.3.0 Copyright 2008-2015 David Alexander, Suyash Shringarpure, John Novembre, Ken Lange Please cite our paper! Information at www.genetics.ucla.edu/software/admixture Random seed: 43 Point estimation method: Block relaxation algorithm Convergence acceleration algorithm: QuasiNewton, 3 secant conditions Point estimation will terminate when objective function delta < 0.0001 Estimation of standard errors disabled; will compute point estimates only. Size of G: 324x13928 Performing five EM steps to prime main algorithm 1 (EM) Elapsed: 0.318 Loglikelihood: -4.38757e+06 (delta): 2.87325e+06 2 (EM) Elapsed: 0.292 Loglikelihood: -4.25681e+06 (delta): 3 (EM) Elapsed: 0.29 Loglikelihood: -4.21622e+06 (delta): 40582.9 4 (EM) Elapsed: 0.29 Loglikelihood: -4.19347e+06 (delta): 22748.2 5 (EM) Elapsed: 0.29 Loglikelihood: -4.17881e+06 (delta): 14663.1 Initial loglikelihood: -4.17881e+06 Starting main algorithm 1 (QN/Block) Elapsed: 0.741 Loglikelihood: -3.94775e+06 (delta): 2 (QN/Block) Elapsed: 0.74 Loglikelihood: -3.8802e+06 (delta): 67554.6 3 (QN/Block) Elapsed: 0.852 Loglikelihood: -3.83232e+06 (delta): 47883.8 4 (QN/Block) Elapsed: 1.01 Loglikelihood: -3.81118e+06 (delta): 21138.2 5 (QN/Block) Elapsed: 0.903 Loglikelihood: -3.80682e+06 (delta): 4354.36 6 (QN/Block) Elapsed: 0.85 Loglikelihood: -3.80474e+06 (delta): 2085.65 7 (QN/Block) Elapsed: 0.856 Loglikelihood: -3.80362e+06 (delta): 1112.58 8 (QN/Block) Elapsed: 0.908 Loglikelihood: -3.80276e+06 (delta): 865.01 9 (QN/Block) Elapsed: 0.852 Loglikelihood: -3.80209e+06 (delta): 666.662 10 (QN/Block) Elapsed: 1.015 Loglikelihood: -3.80151e+06 (delta): 579.49 11 (QN/Block) Elapsed: 0.908 Loglikelihood: -3.80097e+06 (delta): 548.156 12 (QN/Block) Elapsed: 0.961 Loglikelihood: -3.80049e+06 (delta): 473.565 13 (QN/Block) Elapsed: 0.855 Loglikelihood: -3.80023e+06 (delta): 258.61 14 (QN/Block) Elapsed: 0.959 Loglikelihood: -3.80005e+06 (delta): 179.949 15 (QN/Block) Elapsed: 1.011 Loglikelihood: -3.79991e+06 (delta): 146.707 16 (QN/Block) Elapsed: 0.903 Loglikelihood: -3.79989e+06 (delta): 13.1942 17 (QN/Block) Elapsed: 1.01 Loglikelihood: -3.79989e+06 (delta): 4.60747 18 (QN/Block) Elapsed: 0.85 Loglikelihood: -3.79989e+06 (delta): 1.50012 19 (QN/Block) Elapsed: 0.851 Loglikelihood: -3.79989e+06 (delta): 0. 20 (QN/Block) Elapsed: 0.851 Loglikelihood: -3.79989e+06 (delta): 0.00 21 (QN/Block) Elapsed: 0.851 Loglikelihood: -3.79989e+06 (delta): 4.33805e-05 Summary: Converged in 21 iterations (21.788 sec) Loglikelihood: -. Fst divergences between estimated populations: Pop0 Pop1 Pop0 Pop1 0.163 Pop2 0.073 0.156 Writing output files. 会生成两个文件:P,Q
hapmap3.3.P hapmap3.3.Q 1.5 运算admixture时, 添加误差信息
在命令汇总增加一个参数:-B, 速度会变慢喔.
admixture -B hapmap3.bed 3 会生成三个文件:P,Q,Se
1.6 如果你的SNP数据量很大, 跑的很慢
在选择最佳k值时, 可以将SNP分为子集, 比如50k snp分为50个子集, 每个子集1k SNP, 那么根据子集选择最佳K值, 然后根据最佳的K值去跑所有的SNP
1.7 多线程
如果你有多个线程(processors), 可以添加参数-jn, n为线程的个数, 比如你想用4个线程跑:
admixture hapmap3.bed 3 -j 4 2. 参考信息
2.1 如何选择合适的K值
可以同时运行多个程序, 每个程序不同的k值, 比如, 想要k值选择1,2,3,4,5, 可以写为:
for K in 1 2 3 4 5; do admixture --cv hapmap3.bed $K | tee log${K}.out; done 这样跑完之后, 会生成几个out文件,
hapmap3.1.P hapmap3.1.Q hapmap3.2.P hapmap3.2.Q hapmap3.3.P hapmap3.3.Q hapmap3.4.P hapmap3.4.Q hapmap3.5.P hapmap3.5.Q log1.out log2.out log3.out log4.out log5.out 使用grep查看*out文件的cv error(交叉验证的误差)值:
grep -h CV *.out (base) [dengfei@localhost admixture]$ grep -h CV *out CV error (K=1): 0.55248 CV error (K=2): 0.48190 CV error (K=3): 0.47835 CV error (K=4): 0.48236 CV error (K=5): 0.49001 可以看出, K=3时, CV error最小
2.2 如何绘制Q的图表
使用R语言
ta1 = read.table("hapmap3.3.Q") head(ta1) barplot(t(as.matrix(ta1)),col = rainbow(3), xlab = "Individual", ylab = "Ancestry", border = NA) 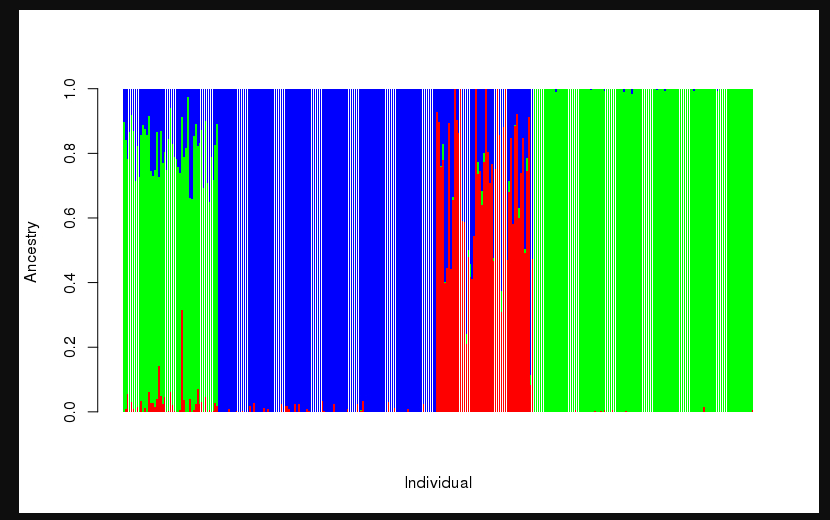
2.3 我需要根据LD去掉一些SNP么?
admixture不考虑LD的信息, 如果你想这么做, 可以使用plink
比如, 这里根据plink 的bed文件进行LD的筛选
plink --bfile hapmap3 --indep-pairwise 50 10 0.1 这里的过滤参数的意思是:
- 50, 滑动窗口是50
- 10, 每次滑动的大小是10
- 0.1 表示R方小于0.1
然后将其转化为bed文件:
plink --bfile hapmap3 --extract plink.prune.in --make-bed --out prunedData 结果输出过滤后的文件为:
prunedData.bed prunedData.bim prunedData.fam 使用过滤后的文件, 从新运行admixture:
for K in 1 2 3 4 5 ; do admixture --cv prunedData.bed $K | tee log${K}.out;done (base) [dengfei@localhost ld-test]$ grep -h CV *out CV error (K=1): 0.52305 CV error (K=2): 0.48847 CV error (K=3): 0.48509 CV error (K=4): 0.49404 CV error (K=5): 0.49828 可以看出K=3, cv error最小, 因此选择k=3
作图:
ta1 = read.table("prunedData.3.Q") head(ta1) barplot(t(as.matrix(ta1)),col = rainbow(3), xlab = "Individual", ylab = "Ancestry", border = NA) 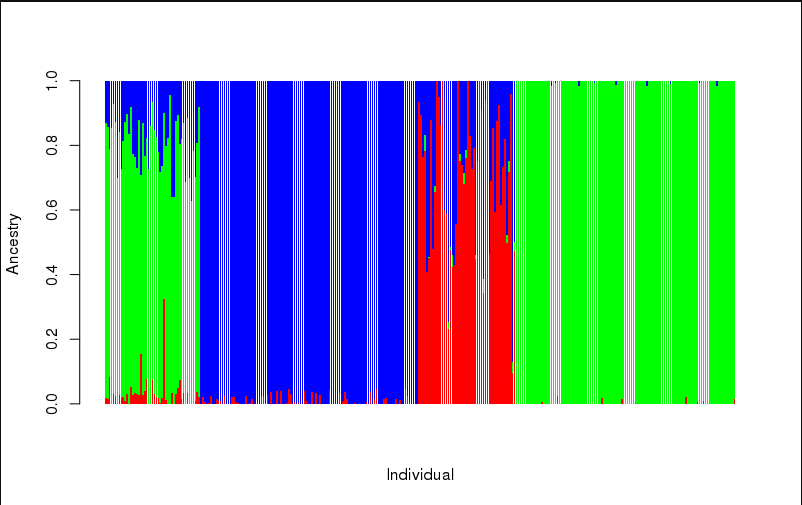
3. 其它
其它见官方的pdf文档
如果您对于数据分析,对于软件操作,对于数据整理,对于结果理解,有任何问题,欢迎联系我。

发布者:全栈程序员-站长,转载请注明出处:https://javaforall.net/199629.html原文链接:https://javaforall.net

![http的幂等性[通俗易懂]](https://javaforall.net/wp-content/uploads/2020/11/2020110817443450-480x300.jpg)
