
大家好,又见面了,我是你们的朋友全栈君。如果您正在找激活码,请点击查看最新教程,关注关注公众号 “全栈程序员社区” 获取激活教程,可能之前旧版本教程已经失效.最新Idea2022.1教程亲测有效,一键激活。
Jetbrains全家桶1年46,售后保障稳定
前两篇介绍了4种靶基因预测软件的下载与安装,以及数据的准备过程。本篇将正式开始进行靶基因的预测, 并对4种个软件的结果进行整理,最终得到4软件结果的交集。
靶基因预测
1、miRanda
miranda file1 file2 [options..]
miranda的使用需要准备两个文件,file1是miRNA序列的fasta文件,file2是mRNA序列的fasta文件。
此外,你还可以根据需求设置可选参数。
以下列举几个常用的参数选项:
-sc S 将得分阈值设置为S [默认:140.0]
-en -E 将能量阈值设置为-E kcal / mol [默认值:1.0]
-strict 严格要求5’种子区配对 [默认:关]
-out file 输出结果到文件 [默认:关]
miranda test.txt total_reverse_CDS201703.txt -out out.txt
grep '>>' out.txt > miranda_result.txt
第一条命令是进行靶基因预测,第二条命令则是从预测结果中提取关键信息(grep搜索含有’>>’的行并输出至指定文件夹)。
2、TargetScan
Targetscan的使用很简单: targetscan_50.pl miRNA.fa mRNA.fa outfile
perl targetscan_50.pl test_targetscan.txt total_reverse_CDS201703_targetscan.txt targetscan_results.txt3、PITA
perl pita_prediction.pl -utr total_reverse_CDS201703.txt -mir test.txt -prefix test &-prefix后面跟的是文件名(不含后缀), 命令后面加个&是为了把程序放在后台运行,因为这个软件运行真的太慢了!
在经过漫长的等待后,程序终于跑完了,结果文件有两个,test_pita_results_targets.tab,test_pita_results.tab,我们要的信息就在test_pita_results.tab文件中,但是这个文件并不是我们真正想要的,PITA这个软件真的太不友好了,还需要我们自己提取△△G小于或等于-10kcal/mol的行.
cat test_pita_results.tab | awk '$13 <= -10 {print $0} > pita_results.txt但是,由于test_pita_results.tab这个文件太大了,我的有400多兆,这条命令执行起来也是超慢的,于是我用了一个很古老的方法,也就是复制粘贴,因为我观察过了,文件中每一条结果是按照△△G由小到大排序的,所以直以将前面小于等于-10的结果(事实上只有很少的一部分)复制粘贴到另一个文本中就好了.
在选择文本时,有一个小技巧.
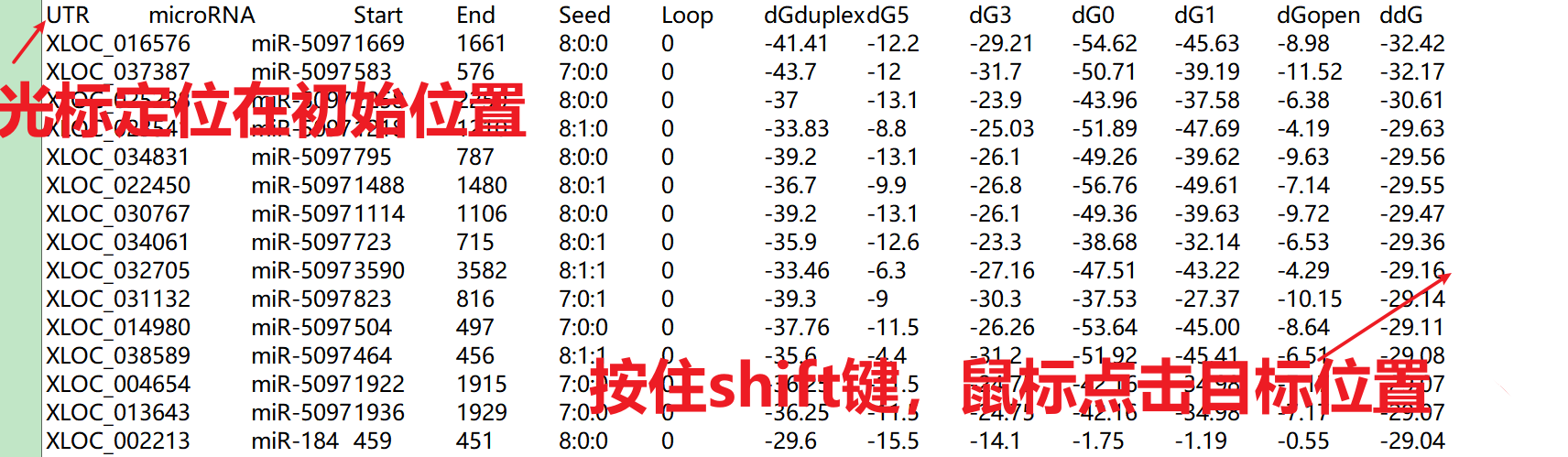

这样就可以快速的选择目标区域了,然后ctrl C ,ctrl V ,搞定!
4、RNA22
将你的miRNA和mRNA文件放在Parameters.properties和RNA22v2.class所在文件夹中,然后打开Parameters.properties文件,根据注释信息修改即可.
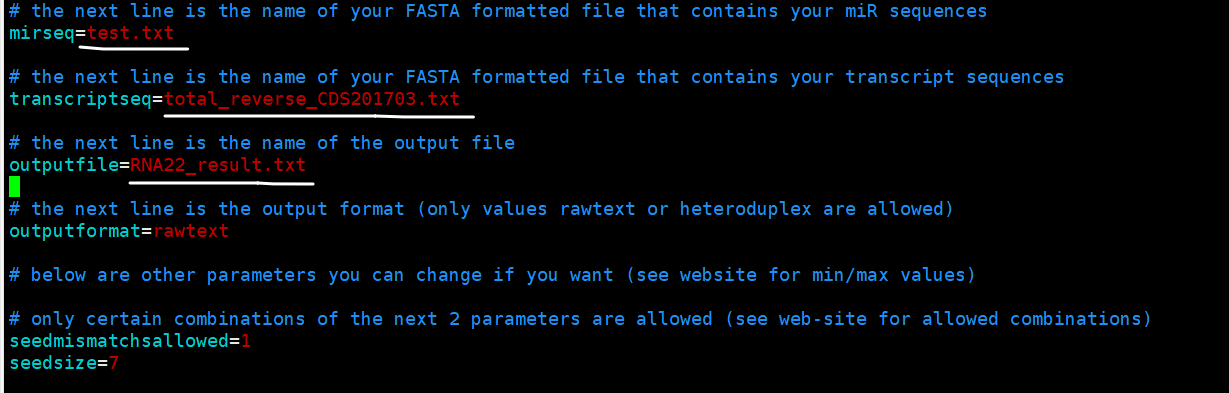
需要java1.6及以上版本才可运行RNA22v2,在服务器输入java –version查看Java版本,若不符合则可下载最新版java.
java RNA22v2因为RNA22的运行也是比较慢的,可以放在后台运行.
结果整理


以上是4种软件靶基因预测结果, miRNA和靶mRNA名称在前两列中, 并且以制表符tab分隔, 我希望从文件中提取前两列的信息,并将其合并为一列.
def TidyMirandaResult(path, inputfile, outfile):
infpath = r'{}\{}'.format(path,inputfile)
outfpath = r'{}\{}'.format(path, outfile)
with open(infpath) as f:
for row in f:
row = row.split('\t')
line = row[0][2:] + ':' + row[1] + '\n'
with open(outfpath, 'a') as r:
r.writelines(line)
def TidyTargetscanResult(path, inputfile, outfile):
infpath = r'{}\{}'.format(path, inputfile)
outfpath = r'{}\{}'.format(path, outfile)
i = 0
with open(infpath) as f:
for row in f:
i += 1
if i == 1:
continue
row = row.split('\t')
line = row[1] + ':' + row[0] + '\n'
with open(outfpath, 'a') as r:
r.writelines(line)
def TidyRNA22Result(path, inputfile, outfile):
infpath = r'{}\{}'.format(path, inputfile)
outfpath = r'{}\{}'.format(path, outfile)
with open(infpath) as f:
for row in f:
row = row.split('\t')
line = row[0] + ':' + row[1] + '\n'
with open(outfpath, 'a') as r:
r.writelines(line)
def main():
path = r'D:\用户\桌面\练习\结果'
TidyMirandaResult(path, 'miranda_result.txt', 'miranda_TidyResult.txt')
TidyRNA22Result(path, 'RNA22_result.txt', 'RNA22_TidyResult.txt')
TidyTargetscanResult(path, 'targetscan_result.txt', 'targetscan_TidyResult.txt')
TidyTargetscanResult(path, 'pita_results.txt', 'pita_TidyResult.txt') # pita结果处理和targetscan是一样的
main()
绘制韦恩图
在线网站:https://bioinfogp.cnb.csic.es/tools/venny/index.html
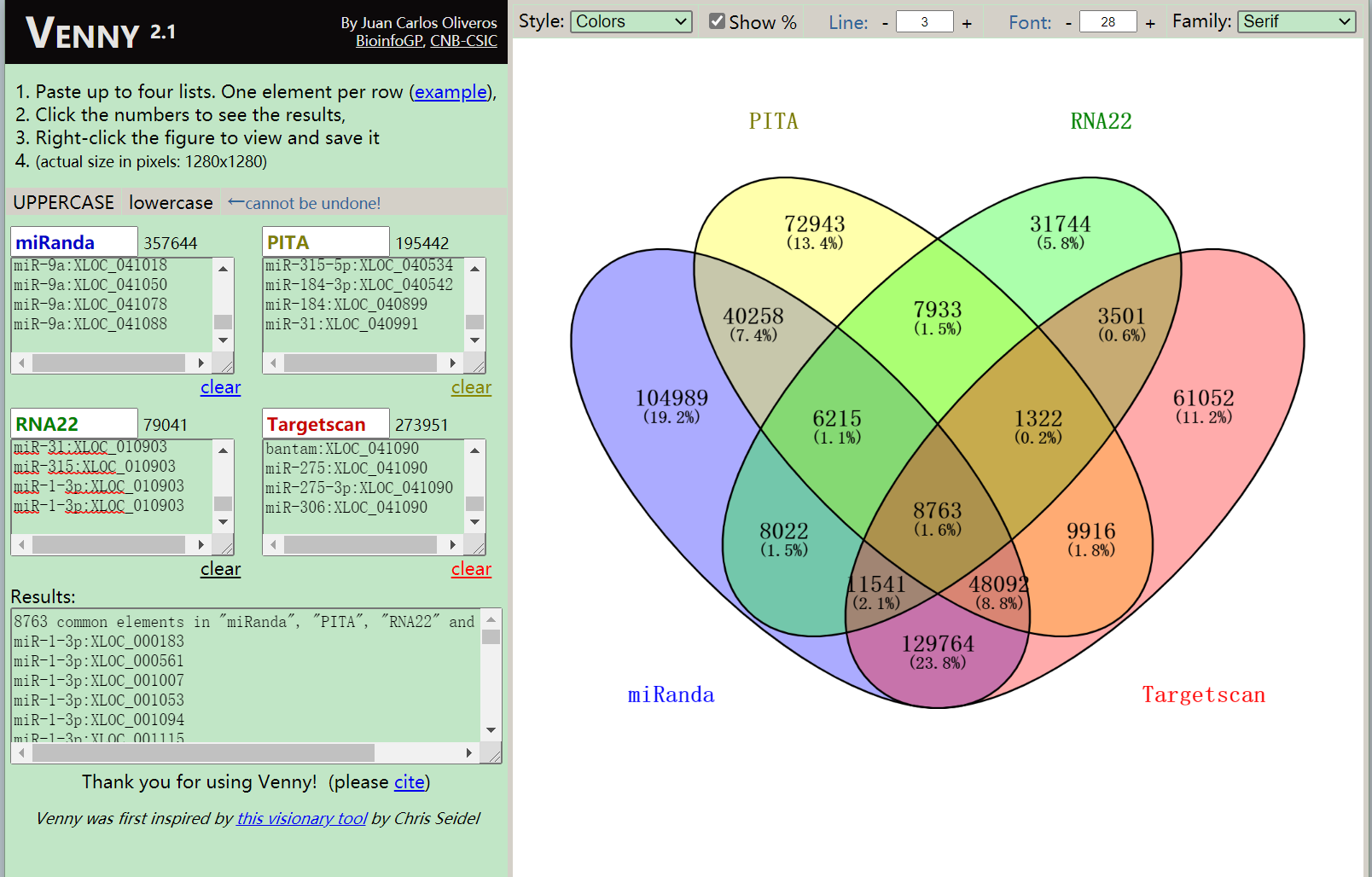
这个网站最多做4组数据的韦恩图,刚好适合我,还可以调颜色,调字体,点击图中的数字就会列出交集结果,个人觉得这个网站做韦恩图还是很方便很不错的.
从结果可以看到,4种软件的交集结果有8763条,意味着测试的miRNA在总转录本中有8763条潜在的靶位点,记住是靶位点,不是靶基因,因为一个基因可能在多个miRNA中有靶位点.
将4软件结果的交集数据保存为txt文本,从该文本中提取出mRNA和靶基因名称
i = 0
with open(r'D:\用户\桌面\练习\结果\4软件结果交集.txt') as f:
for row in f:
i += 1
if i == 1:
continue
row = row.split(':')
line = row[1]
with open(r'D:\用户\桌面\练习\结果\靶基因.txt', 'a') as r:
r.writelines(line)有了靶基因名称就可以做一些KEGG富集分析了.
发布者:全栈程序员-站长,转载请注明出处:https://javaforall.net/213952.html原文链接:https://javaforall.net


